Are you ready for Brexit? Do you have a Brexit contingency plan?
You may need either an EU/EC European authorized representative based in EU-27 countries or a UK authorised representative (so-called "UK Responsible Person") based in UK, or may even need both EU & UK representatives, depending on different brexit scenarios.
Register/Notify your MD-Medical Devices & IVD-In Vitro Diagnostic Medical
Devices with MHRA in UK & other EEA (EU/EFTA) authorities by world-leading
consultancy- Wellkang team based in both UK (England) & EU-27 (Ireland).
Wellkang team can help you under all Brexit scenarios!
Click here to get FREE Guide Now! |
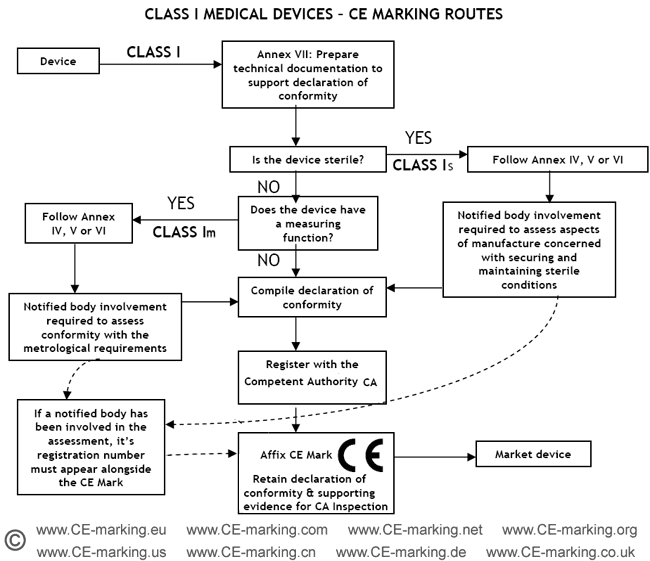
Steps for Class I medical devices compliance
- Classification: ensure the device is a Class I medical device.
- Choose Conformity Assessment Route: refer the flow chart below.
- Compile the Technical File.
- Declaration of Conformity.
- Appoint an Authorised Representative. (register with the Competent Authority)
- Vigilance and Post Market Surveillance. (affix CE marking & market the products)
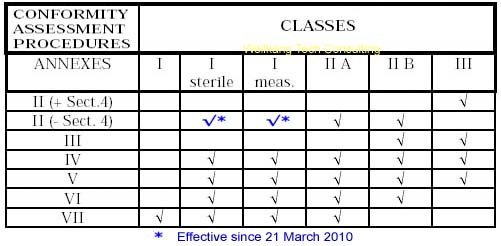
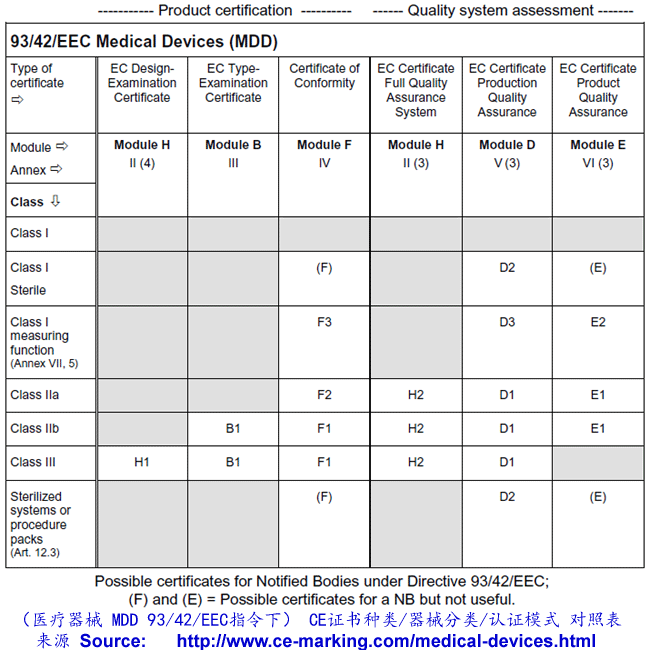
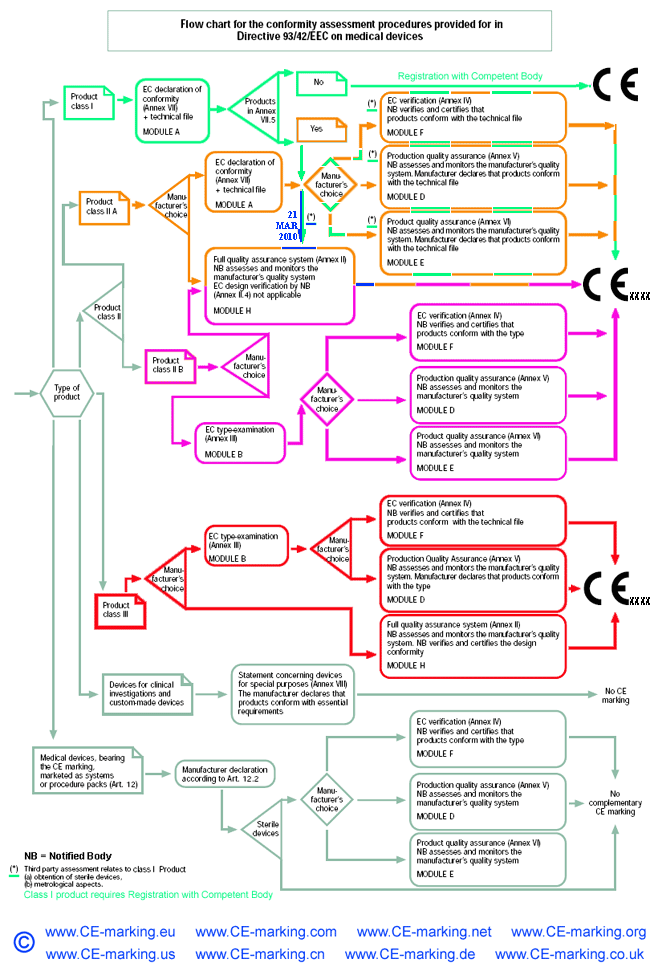
Class I Medical Devices: Conformity Assessment Routes
The conformity assessment routes for Class I Medical Devices
- The manufacturer is responsible for ensuring that his product complies with all the
relevant Essential Requirements of the Directive and must draw up a written statement to
this effect (self-declaration).
- Class I Medical Device without a measuring function and supplied in non-sterile condition does NOT require the involvement of a Notified Body. Conformity to the International and European Standard EN ISO 13485 is voluntary.
- Manufacturers of sterile products and devices with a measuring function must apply to a Notified Body for certification of the aspects of
manufacture relating to sterility or metrology.
- Once the manufacturer is satisfied that his products meet all the relevant Essential Requirements, the manufacturer, or his European Authorised Representative if the manufacturer is located outside of EEA, must register with the Competent Authority (CA). He may then affix the CE marking on his products and place them on the EEA market.

FAQ/Q&A: Questions and Answers about CE Marking of Medical Devices
About CE Marking:
| Home | About Us | Contact Us | Copyright & Disclaimer | Privacy | Log-in | Order Now | |